
Phillip Compeau, Ph.D.
Teaching Professor
Asst Dean for Innovation in Computing Education
Computational Biology Department
Carnegie Mellon University
SARS-CoV-2 Software Challenge: Evolutionary Trees and Multiple Alignment
by Kunal Joshi, Maggie Zhang, & Phillip Compeau
Introduction
In the previous assignment, we demonstrated how to assemble and annotate the SARS-CoV-2 genome that caused the COVID-19 pandemic. Although knowing the genomic identity of this virus is very important in order to compare it to other coronaviruses (most notably the SARS-CoV virus that caused the 2003 outbreak of SARS), researchers are interested in determining the genome of thousands of different individual viruses as the virus spreads around the planet.
This idea of viral genomic surveillance is not new; it became famous in 2014 when researchers captured patient samples to identify the source of the Ebola outbreak. In the case of SARS-CoV-2, we know that the virus likely originated in Wuhan, but genomic surveillance helps us determine how the virus is mutating in human hosts as it spreads and to identify new variants of the virus for further study, as well as whether vaccines work on these variants.
The computational task at hand is to construct an evolutionary tree, along with a multiple alignment, of viral genomes sampled from many patients. Such an endeavor requires sophisticated algorithms that are efficient enough to handle many 30,000 base pair genomes.
We will be studying samples taken from patients in the United Kingdom (UK), not because the virus there has an affinity for milky tea, but because their reliance on a nationalized health service means that they have collected and published more viral genome data than we have in the US.
In this assignment, we will place ourselves in the shoes of researchers sequencing genomes and see if we can find any interesting variants within the patient viral genomes. Are the variants we find changing over time?
Sampling and aligning patient genomes
To examine how SARS-CoV-2 is mutating over time, we collected a random sample of 100 coronavirus genomes taken from patients in the UK every two weeks from November 2020 to March 2022 by consulting the COVID-19 Genomics UK Consortium. We will provide a subset of genomes from this consortium as part of this challenge, but if you’d like to explore the full data for yourself, the consortium provides direct downloads to both metadata as well as all genomes stored on their site. (Warning: these files are very large.)
To analyze the genome data, we will perform a multiple sequence alignment of each file, which will reveal possible mutations in the genomes. In this assignment, we will use Clustal, a landmark algorithm that produces an evolutionary “guide” tree in order to produce a multiple sequence alignment. Clustal is a heuristic, meaning that it may not produce the optimal alignment solution but is reasonably efficient.
The most up-to-date, optimized version of Clustal (known as Clustal Omega) still takes approximately two hours to align 100 coronavirus genomes, which explains why we sampled only 100 samples on each day instead of 1000.
Click here to download our data for this assignment. You will see that each folder in the dataset has the format “YYYY-MM-DD” and contains two .fasta files (we introduced this file format in the assembly/annotation assignment). The file YYYY-MM-DD.fasta contains the 100 genomes sampled on this day, and the file YYYY-MM-DD_A.fasta contains a pre-processed alignment file that we will discuss soon. Feel free to take a peek at a genome file using a text editor and see the genomes.
It is outside the scope of this assignment, but if you are interested in running the alignment algorithm on each sample yourself, you can do so by using the European Bioinformatics Institute’s free Clustal tool, or by running Clustal on a Galaxy server (warning: Galaxy has frequent timeouts on large jobs), or by downloading a program that has Clustal built into it, such as MEGA.
Finding mutations
To view our viral genome alignments, we will use a specialized tool. The National Center for Biotechnology Information (NCBI) provides a great MSA viewer at the link below.
https://www.ncbi.nlm.nih.gov/projects/msaviewer/
Visit this link, and upload the alignment file from 2020-11-16 (2020_11_16_A.fasta). You can do this by clicking on the Upload button, selecting Data File from the sidebar menu, and following the upload instructions. Then click Close when the data has been uploaded.
Once completed, the viewer should open, as shown in the screenshot below.
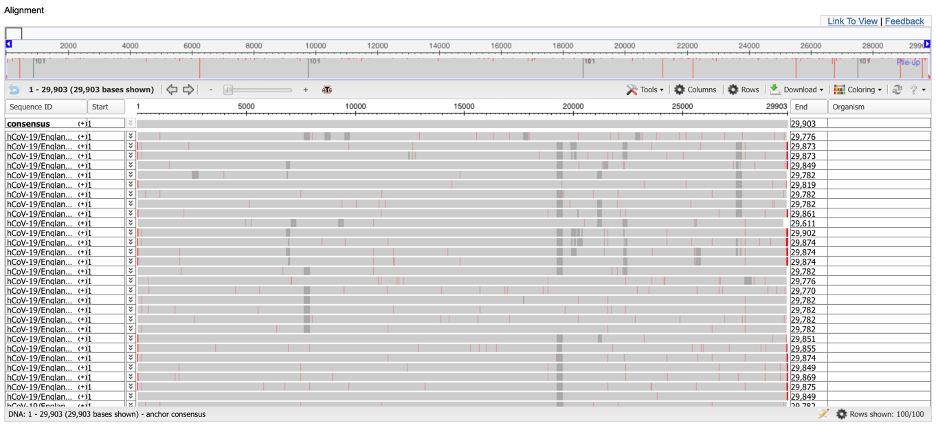
We cannot see individual nucleotides currently because the viewer is presenting an eagle-eye view of the alignment. You can obtain more information about a given position by clicking on it. You can also use the zoom feature at the top of the page to focus on a smaller region of the alignment.
Exercise 1: All 100 genomes have the same substring of five nucleotides ranging from column 2251 of the alignment to column 2255. What is the substring?
Exercise 2: What do the red vertical bars signify in the multiple alignment? What about the gray regions? (Hint: You may find IUPAC notation helpful.)
Exercise 3: Are there any regions of the genome that you find particularly interesting in terms of studying viral variation? Justify your answer.
Exercise 4: If you look at the ends of the alignment, you will see that there is a lot of variation at the ends of the alignment. Why do you think this is? (Hint: this is a question about genome assembly in disguise; take a look back at the contig that we produced in the assembly assignment if needed.)
Now that we have explored the alignment data for 2020-11-16, let’s check in on the virus a few weeks later. Follow the same steps to view the alignment for 2020-12-28 (2020_12_28_A.fastaa
Exercise 5: Use the notion of a molecular clock to explain why it is not surprising to see an increase in the number of viral mutations over time.
What is worrisome about the increased number of mutations that you can see in the alignment is not that mutations are occurring, but that their frequency appears to be increasing in the same column. This is not what we would expect if mutations are simply occurring randomly, and it implies that the virus may be gaining mutations that are producing variants that are in some way “more fit”.
Furthermore, the variability seems to change as we pass over the alignment. One of the more variable regions corresponds to the spike protein, one of the genes that we identified when annotating the SARS-CoV-2 genome in the previous challenge. In the 2020-11-16 alignment, this gene occurs starting at column 21,563 (if you zoom in on the alignment, then you will see an “ATG” at this position, which is a start codon). We will focus in on the spike protein, and discuss it exclusively in the next challenge, because the spike protein binds to the ACE2 enzyme on the surface of human cells; minor changes in bonding affinity can therefore have enormous consequences on the infectiousness of the virus.
We will now take the time to discuss some of these mutations in greater detail.
Profiling individual mutations in the alpha variant
In this section, we will give an overview of three mutations that researchers identified in the spike protein gene occurring together in many sampled viruses from November 2020. These mutations, taken together, are some of the mutations that defined the first main variant of SARS-CoV-2, called B.1.1.7 or the alpha variant.
The three mutations are called N501Y, ΔH69/V70, and P681H. To explain this shorthand, N501Y means that the 501st codon of the spike protein changed from encoding an N to encoding a Y, and P681H means that the 681st codon of the spike protein changed from encoding a P to encoding an H. As for ΔH69/V70, this shorthand indicates that the 69th and 70th codons of the spike protein are deleted.
N501Y (the ACE latcher)
This is a well-studied single-nucleotide mutation first discovered in April 2020; it allows the coronavirus to more tightly fit the ACE2 receptor on human cells (the receptor that facilitates the virus’s cell entry).
Because the spike protein begins at position 21563 of both the 2020-11-16 alignment and the 2020-12-28 alignment, this mutation occurs at position 23063 of the alignments.
Exercise 6: At the level of nucleotides, what was the original mutation? How many of the 2020-11-16 genomes have the mutation, and how many of the 2020-12-28 genomes have the mutation?
ΔH69/V70 (the antibody butter)
This deletion is present in several coronavirus variants and is therefore referred to as a recurrent deletion region. Although studies have confirmed this mutation does make the coronavirus more infectious, scientists are not entirely sure why. They speculate it may prevent antibodies from binding as tightly.
This mutation removes the 69th and 70th amino acid of the spike protein, which corresponds to positions 21765 through 21770 (inclusively) of both the 2020-11-16 and 2020-12-28 alignments.
Exercise 7: What is the nucleotide sequence that is deleted? How many of the 2020-11-16 genomes have the mutation, and how many of the 2020-12-28 genomes have the mutation?
Exercise 8: You know that just because we see gap symbols does not mean we can infer that a deletion occurred. It may be that these six nucleotides were inserted in an ancestral sequence. Use the 2020-11-16 alignment to argue why the mutation is most likely a deletion.
P681H (the enzyme booster)
This mutation is also present in many coronavirus lineages internationally. Scientists believe that this mutation makes it easier for human enzymes to prepare the spike protein for cell entry.
The mutation occurs on the 681st amino acid of the spike protein and at nucleotide position 23604 of our alignments.
Exercise 9: What was the original nucleotide at this position? How many of the 2020-11-16 genomes had it? What was the mutation, and how many of the 2020-12-28 genomes had acquired the mutation?
Identifying variants and comparing variant prevalence over time
B.1.1.7 was just the first of many different SARS-CoV-2 variants to make headlines during the pandemic. In what follows, we will discuss how we can differentiate SARS-CoV-2 variants based on mutations in their spike proteins and then transition to track these variants over time in our sampled UK genomes.
Exercise 10: Explain how we might write a function that, given a SARS-CoV-2 genome, returns the sequence of the spike protein in the genome.
You may also be wondering: how can researchers identify variants in the first place? We should find a computational approach that is superior to hunting through alignments of thousands of coronavirus genomes to find correlated mutations by visual inspection. It would be nice, for example, to “cluster” coronavirus genomes into groups, and then assign each cluster to its own variant.
To cluster viruses, we could determine a “distance” between every pair of genomes in our dataset, and then assign viruses to n-dimensional space so that the distances between points in this space approximate the “distances” between the corresponding genomes. One way of comparing a pair of viruses would globally align either their entire sequence or that of their spike proteins, but scaling this approach for all pairs of thousands of genomes may prove inefficient.
Instead, for a fixed (small) value of k, we could form a frequency map for each genome that assigns each k-mer in the genome to its number of occurrences in the genome. For example, the following figure shows the frequency maps of the two strings ACGTATACACGTAT and TATCGGTATATCCTAC.
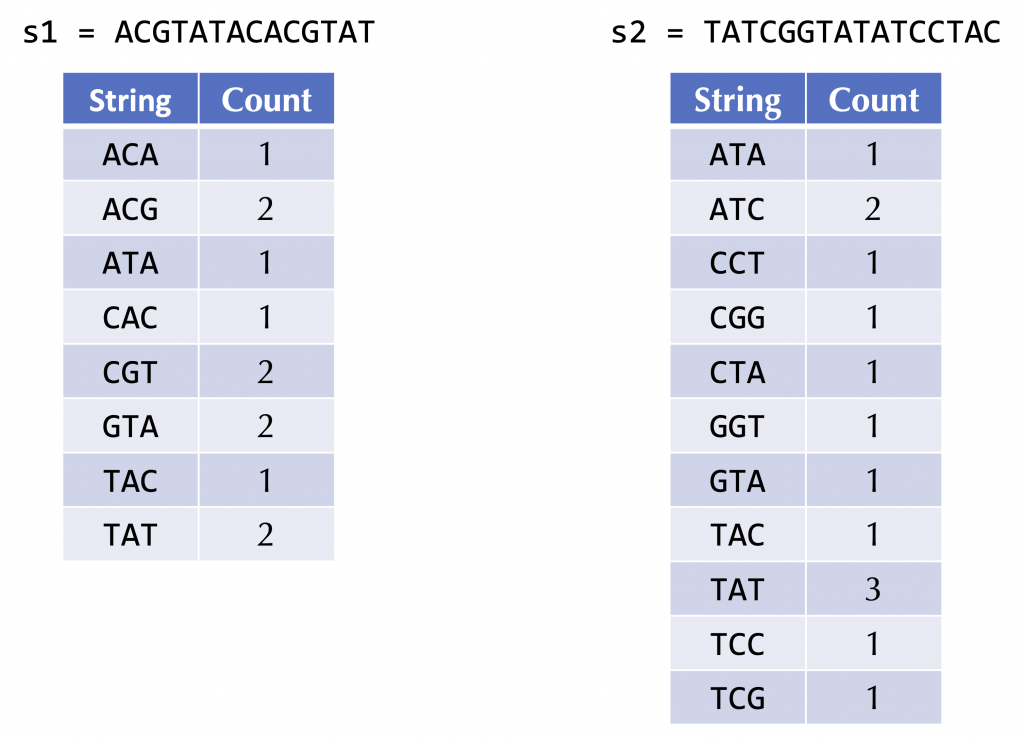
Exercise 11: Outline an approach that, given the frequency maps of two strings, defines a “distance” between the two strings. Explain how your approach would quantify the distance between the above two strings.
After we can identify variants, the next problem of interest is how to classify a newly sequenced viral genome according to its variant (i.e., determining the “cluster” to which it belongs). Once researchers have identified variants, they look for what characteristics genomes from the same variant tend to share. As a result, researchers have built a database cataloging each known frequent mutation along with its prevalence in each variant, which can be explored at the following link: https://covidcg.org/?tab=report.
We are interested in exploring the following three very prevalent variants that arose in the human population: B.1.1.7 (“alpha”), B.1.617.2 (“delta”), and BA.1 (the first variant known as “omicron”). After following the above link, ensure that each of these variants is present in a column and scroll down to explore the mutations shown under “S”, which are sorted according to their codon. For example, you will see that the N501Y mutation that we previously identified as an alpha variant mutation is also present in BA.1 (as well as the variant B.1.351). As a result, it might not be the best course of action to use this mutation in order to differentiate alpha from other variants, since a significant number of viruses classified as the BA.1 variant possess it as well.
Exercise 12: There is one spike mutation that is common to every variant shown in the table at 100%. What is it?
Unlike N501Y, we would like to pick a small set of mutations that completely differentiate the three main variants from each other. To do so, we will select A67V (which occurs only in BA.1), A570D (which occurs only in B.1.1.7), and P681R (which occurs only in B.1.617.2). A virus containing one of these mutations will be classified according to the main variant that possesses that mutation, and a virus containing none of these mutations will be classified as “other” (perhaps belonging to the original SARS-CoV-2 strain).
Exercise 13: Give two additional spike protein mutations that are exclusively contained by B.1.1.7 viruses.
We now are prepared to identify the changes in frequencies of SARS-CoV-2 variants over time.
Exercise 14: The next strain to arise in the UK was B.1.617.2 (“delta”). How many of the 100 genomes for 2021-05-17 corresponded to this variant?
Exercise 15: After delta came omicron (whose first strain was BA.1). How many of the 100 genomes for next strain to arise in the UK was B.1.617.2 (“delta”). How many of the 100 genomes for 2021-12-13 corresponded to this variant?
The previous two exercises are nice, but we would like to automate our classifier so that it can quickly classify all genomes in our dataset.
Exercise 16: Write a function to excise the spike protein from a given coronavirus genome. Then, apply this function to every genome in our dataset (you may not successfully excise every genome). After doing so, label each genome according to one of the three variants above, or “other”. Then, provide a bar chart that shows the percentage of each variant for each sampled date.
(Hint: in BA.1, A67V will occur starting at the 199th nucleotide of the spike protein; in B.1.617.2, P681R will occur starting at the 2041st nucleotide of the spike protein; and in B.1.1.7, A570D will occur starting at the 1699th nucleotide of the spike protein because the 69th and 70th amino acids are typically deleted in this strain).
Looking ahead
In this section, we have used multiple alignments and evolutionary tree construction to help identify and differentiate variants. But it is still unclear how changes in SARS-CoV-2, and in particular its spike protein, have an effect on the function of that spike protein and the behavior of the virus.
In the third SARS-CoV-2 challenge, we examine the structure of this protein and examine how it differs from the spike protein of SARS-CoV, the virus responsible for the 2003 outbreak.